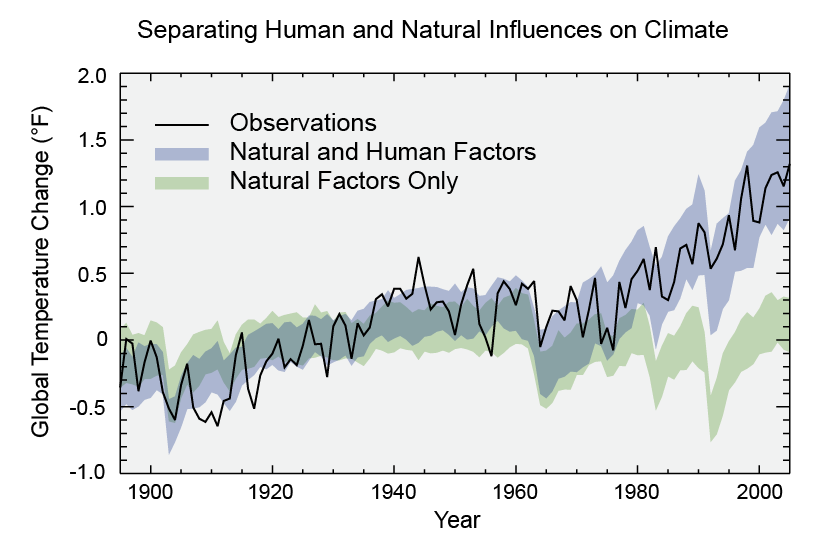
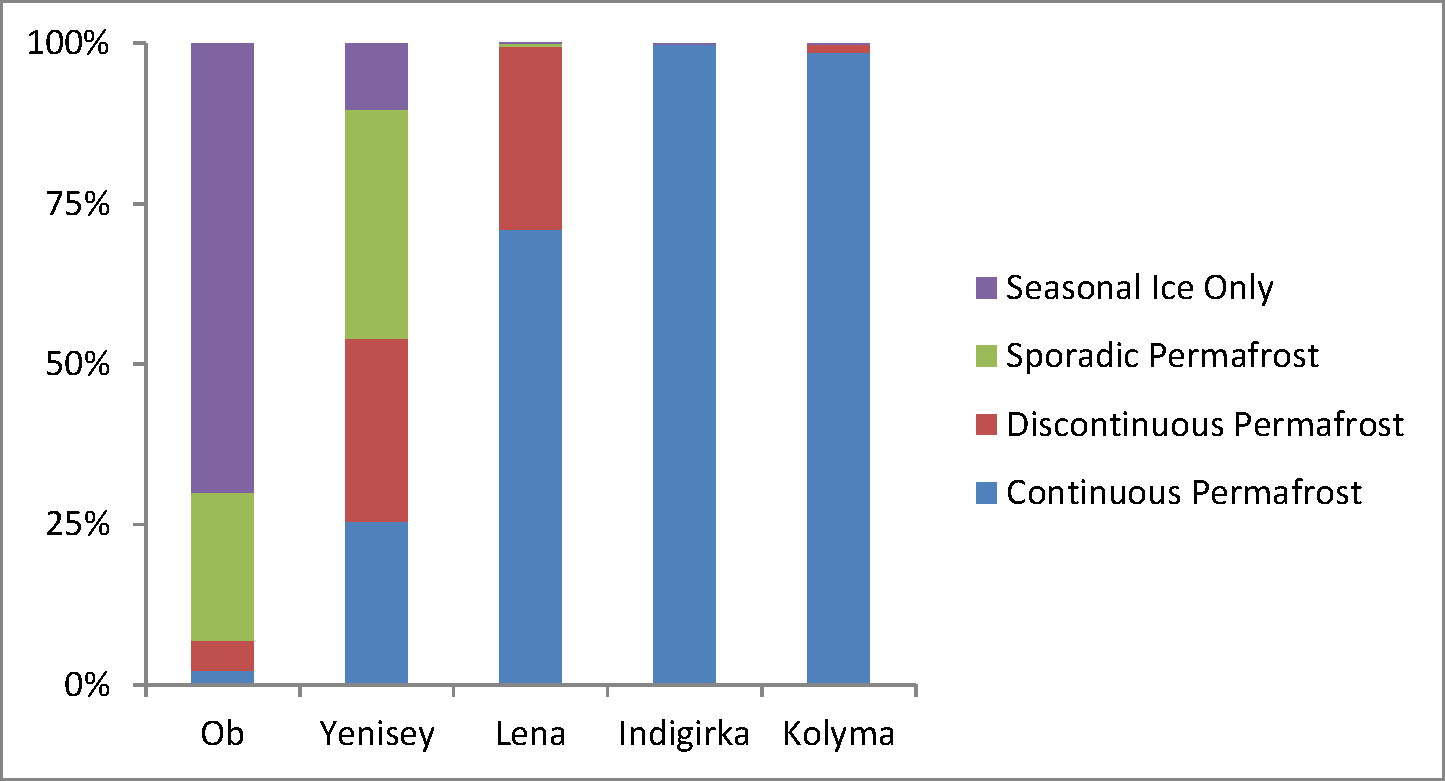
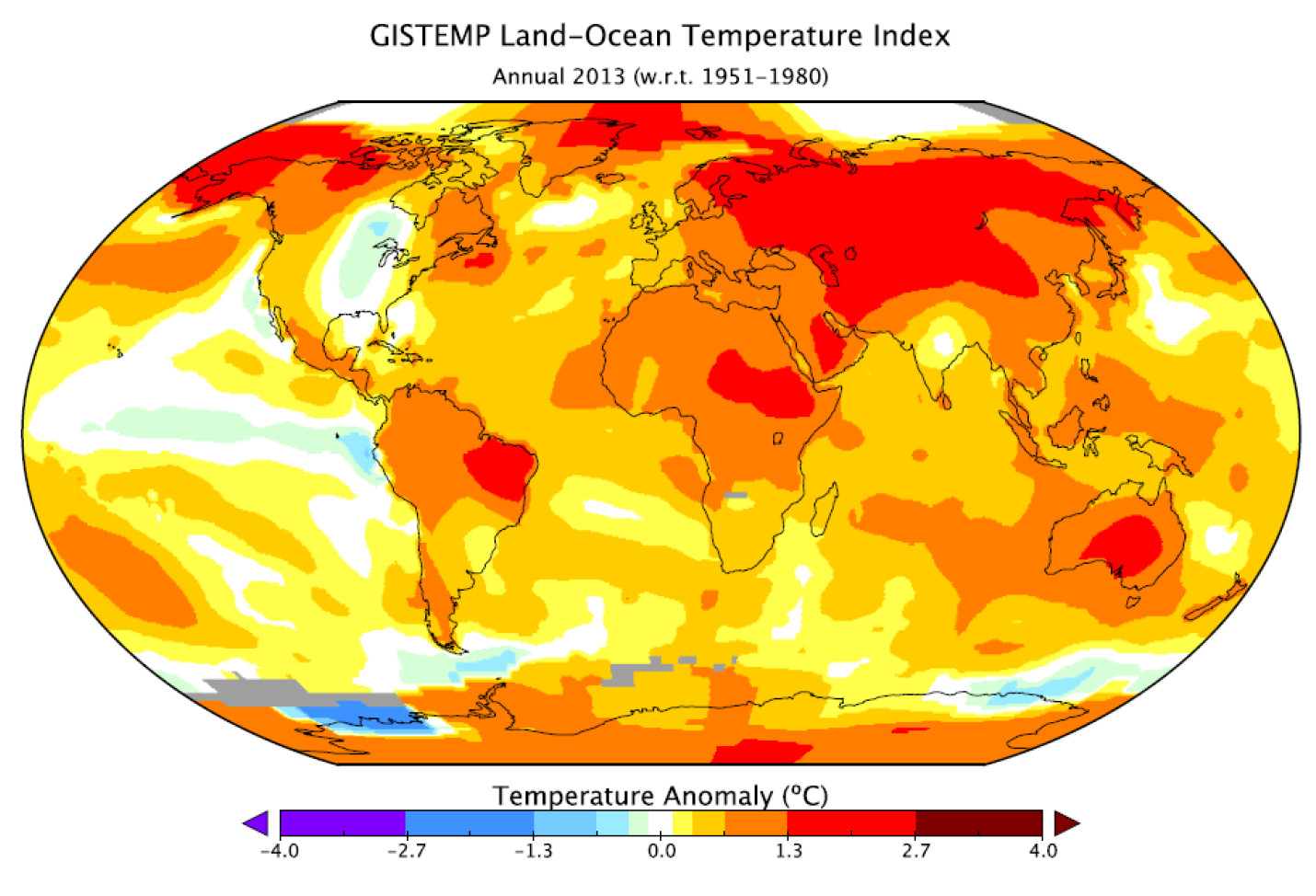
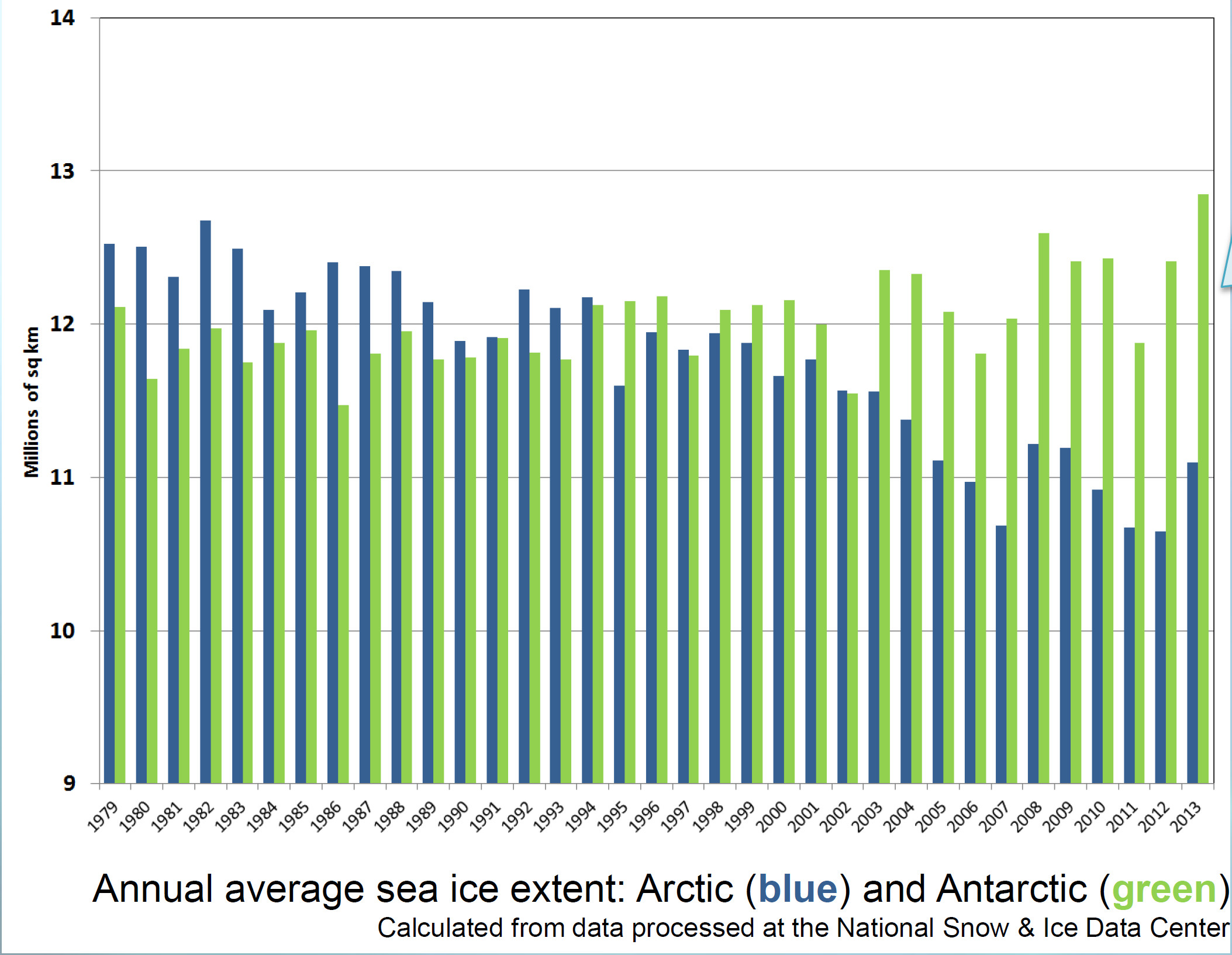
A new report from Yale has found that “belief” in climate change among the American public is decreasing, and that now less than half of people believe that climate change is man-made.
So why is this? My personal guess is that we’ve all got a little bit of David Cameron inside us, and when things got tough we decided to “cut the green crap”. Shrinking fincances have led to a more selfish outlook, we just can’t justify spending more on sustainable energy when there are other, more basic, needs to consider. Couple this with a natural desire to believe that whatever bad things happen are not our personal fault, and an increasingly vocal climate skeptic lobby, and it’s understandable that people would want to switch sides.
But wait, burying our heads in the sand is not the answer. Things have changed recently, measurements have been taken and trends have been spotted that, with the right spin on them, would appear to play right into the hands of the global warming deniers, yet the reality is that we should be as concerned as ever about the future of our planet. Recently I have spoken to several well-educated, some extremely well-educated, people who, despite all the coverage are unconvinced that we, you me and 7 billion other human beings, are responsible for the changing climate, or even that it is changing at all. This seems to be a new thing, a few years ago they might have been on the other side of the debate. Has climate skepticism become fashionable?
I’m not about to start presenting all the data and rebuking every argument about climate change, the volume of data is too large and there are so many points to argue about that I don’t have the time. However, lots of other people do have the time:
The latest IPCC report is probably a good start if you want the “official” summary:
http://www.climatechange2013.org/images/uploads/WGI_AR5_SPM_brochure.pdf
A more targeted and less hardcore site goes through the climate skeptic arguments one by one:
http://www.skepticalscience.com/
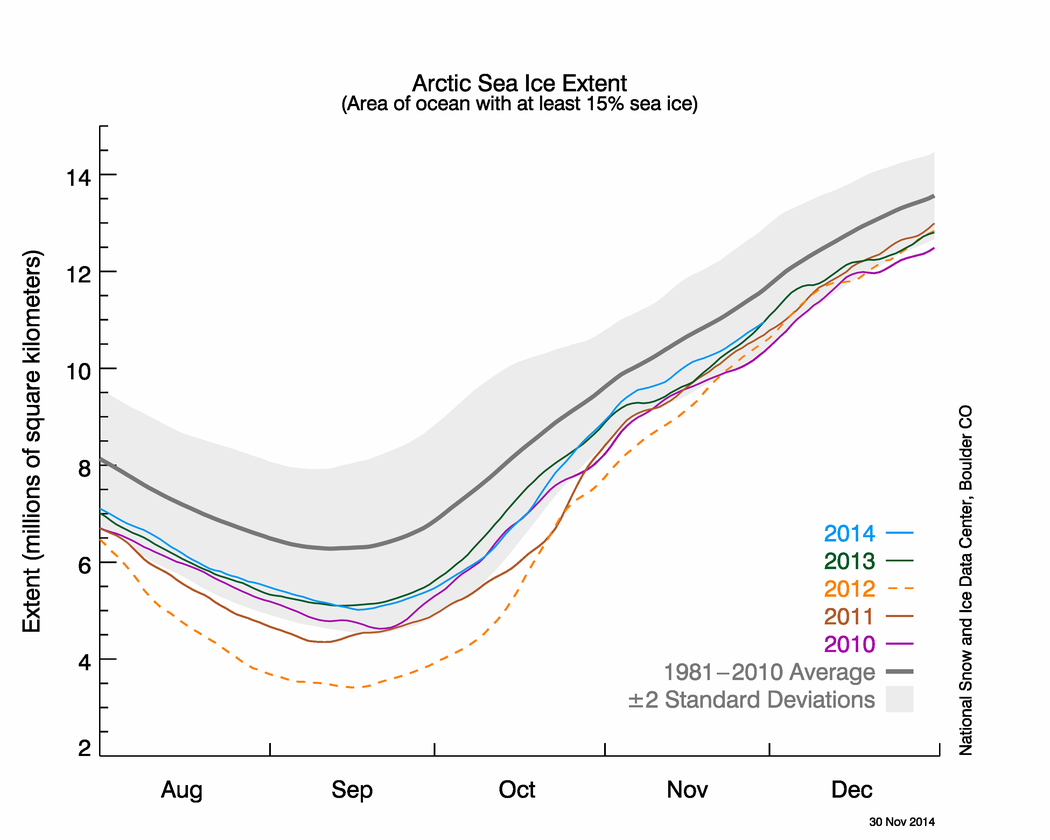
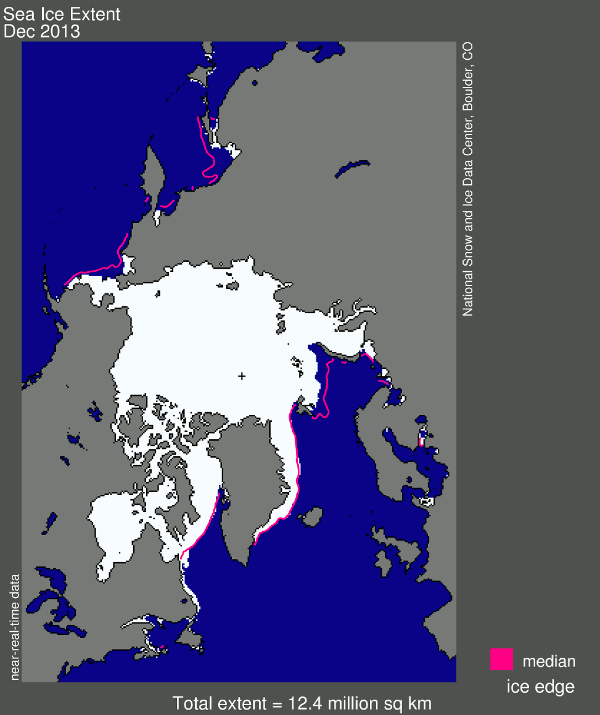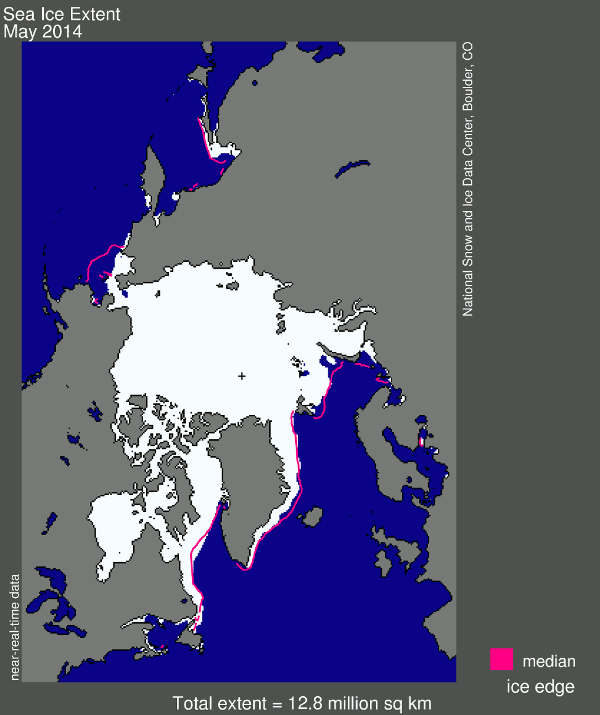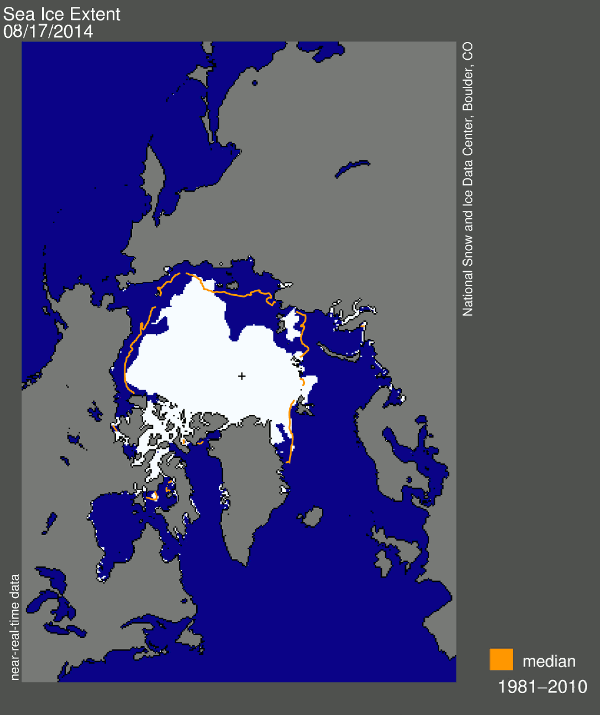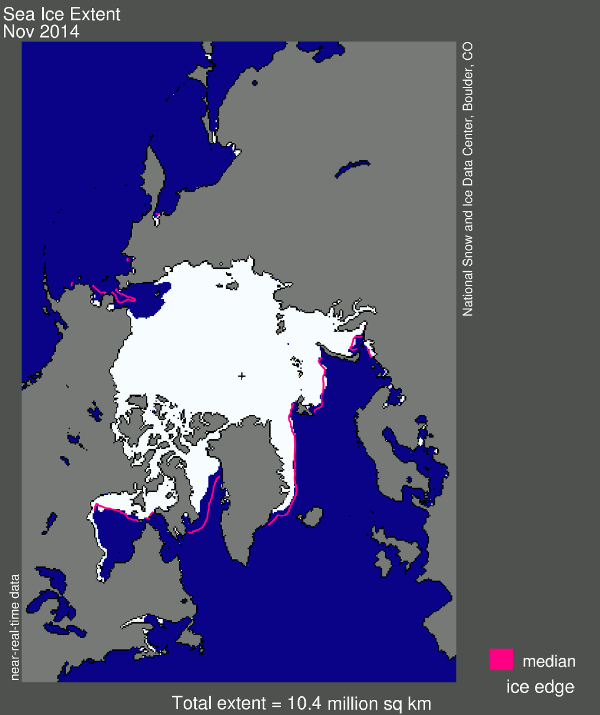
And now for my two-pence worth. It’s mostly an appeal for common sense, from both sides of the argument. Journalists are always keen to get a good story, and people with opinions are always keen to emphasise evidence that they are right. However, our planet is a complex system and a single piece of evidence does not sway things. 2012 was a record-breaking year for ice cap retreat in the Arctic, the summer ice coverage was the lowest ever. This led to a lot of reporting which, justifiably, picked up on this and suggested that it might be a bad thing. But did they go too far? Was one data point enough to justify widespread panic? Probably not, but it was something to bear in mind and consider along with all the other available evidence. Subsequently, since we are not yet in a run-away global warming apocalypse, when 2013 failed to break the record again and was merely in line with other data from the 2000s, the journalists on the other side of the argument got to crow that the world was cooling down again. Now they’re almost certainly wrong in every degree – a quick look at longer-term trends would show that 2013 was still far lower than the average and that ice thickness is reducing quickly – but both scientists and science journalists must beware of crying wolf based on a single year’s data.
Getting people worried about climate change was a good thing, but doomsday prophesies about massive immediate changes that have not been borne out will only lead to disbelief. Just like the size of the ice cap, public belief in global warming is probably due to a number of nebulous factors and might fluctuate from year to year and we as a scientific community must be sensible about the way in which our concern is projected in the media, concentrating on boring but incontrovertible long-term trends rather than sexy but one-off events. As ever, climate != weather.